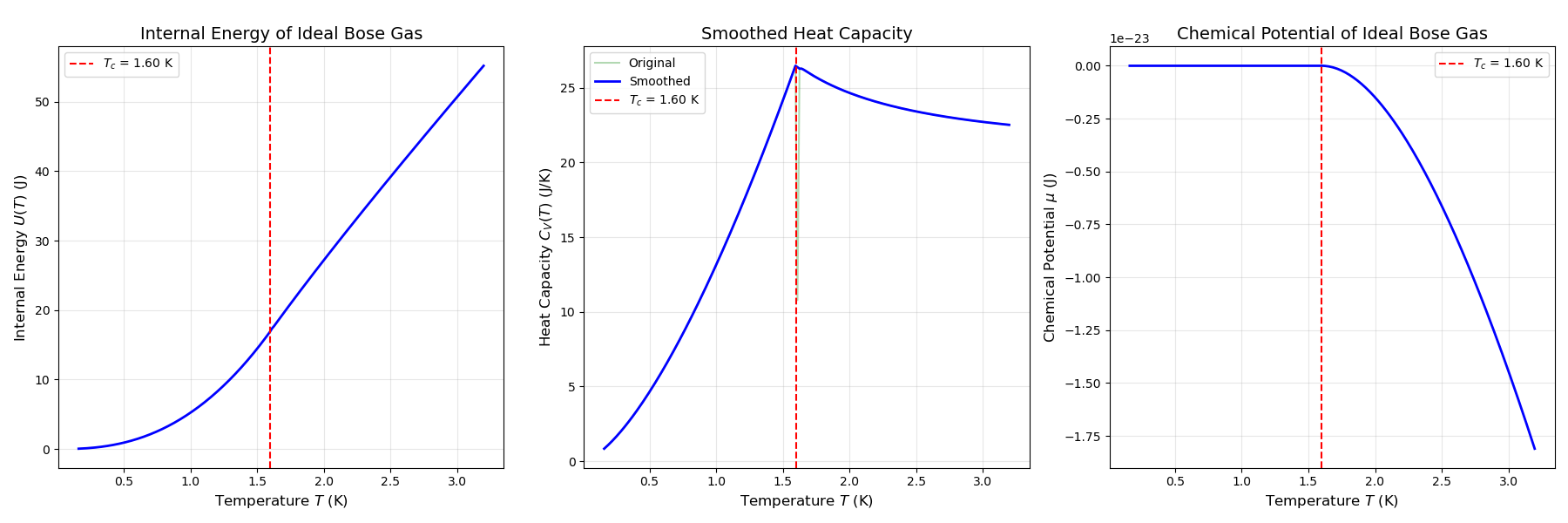
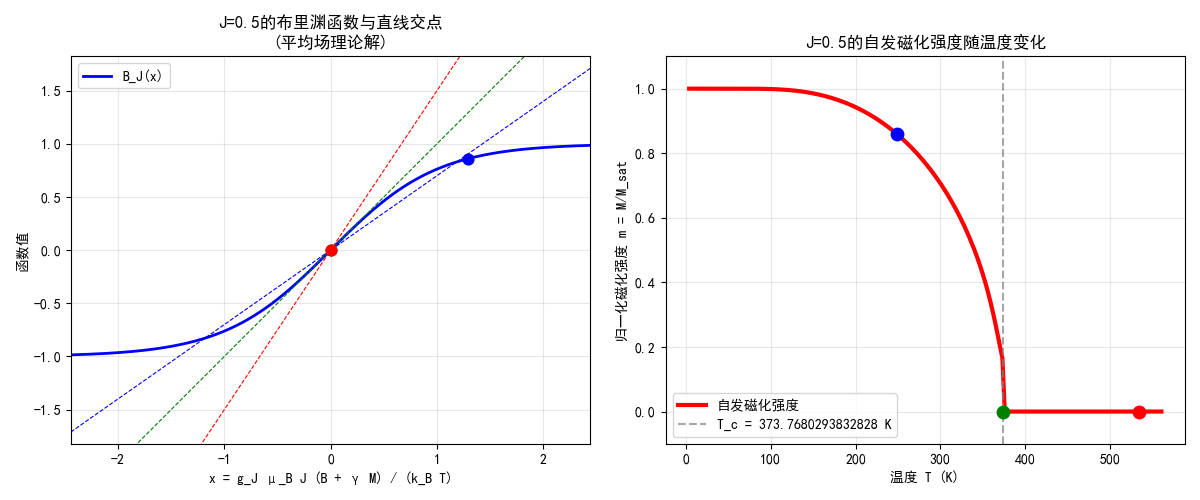
系综理论
相空间和刘维尔定理
最概然分布的方法处理近独立的粒子,但是不能处理有强相互作用的粒子,这时候就要请出平衡态统计物理的普遍理论——系综理论。
在经典力学中,N个全同粒子(每个粒子的自由度为r)的总自由度为$f=Nr$。那么可以通过广义坐标$q_i,i=1,\cdots,f$和广义动量$p_i,i=1,\cdots,f$构成2f维度的相空间。系统的运动状态由哈密顿正砸方程决定,即
对于保守系统,哈密顿量就是系统的能量,即
这说明系统其实是在像空间的2f-1维曲面上运动的。系统在t时刻处于某个点$(q_1,\cdots,q_f,p_1,\cdots,p_f)$,如果我们有N个系统,那么对描述系统分布的密度函数$\rho(q_1,\cdots,q_f,p_1,\cdots,p_f,t)$积分,有:
系统的密度分布函数同样有运动方程,即
其中
刘维尔定理指出:
证明:
刘维尔定理指出系统的分布函数是不随时间改变的。
系综理论
统计物理的发展初期,人们主要通过力学来推导具有统计意义的热力学量,比较著名的是气体分子动理论(你肯定见过从动量交换的角度推导出气体压强)。以玻尔兹曼和麦克斯韦为首的物理学家希望从时间平均上求解系统的宏观物理量$B(t)$:
这里的$\tau$取一个宏观短微观长的时间间隔。宏观短是因为避免宏观物理量发生可以观测的变化,微观长是因为希望平均掉微观物理量的涨落。
这一表达式除了定义外,没有任何的作用——粒子数($N\sim 10^{23}$)和时间($\tau$导致的步数视系统的微观频率而定)实在是太大了。不过现代的算力支持第一性原理的分子动力学模拟,从中可以得到系统的时间平均。
必须指出的一点是,即便是孤立系统,也并非完全孤立:微弱的引力场和电磁场,乃至真空本身都会与其发生相互作用。所以对于孤立系统,我们也要求一个宏观短的时间,尽管他的跃迁不会很大。
吉布斯等人提出使用系综平均来等效代替时间平均(假设1)。系综平均是指在一个系综中,所有可能的体系对应的微观物理量都被加权考虑,权重就是该体系可能的概率:
这时候,我们只需要关心两件事情:微观物理量$B(q,p)$和分布函数$\rho(q,p,t_0)$。一般来说,微观物理量视系统的性质而定,而分布函数其实具有统计上的普遍性。
所谓系综理论,就是研究分布函数随系综性质的变化规律。平衡态条件对系综理论的帮助是,平衡态下的宏观物理量时间不变,意味着分布函数是时间无关的,即:
对于假设1,即时间平均是否等于系综平均,物理学家们最初的Argue是遍历假说,这一点在数学上可以导出假设的正确性。然而,遍历假说并不适用于所有的系统。这也是统计物理学家抛弃纯力学原理,转而通过统计上的一些假设(后验正确的)来建立统计物理学的原因之一。
微正则系综
等概率原理
为了求解一个具有确定的粒子数$N$、能量$E$和体积$V$的孤立系统(称之为微正则系综),我们会假设等概率原理(假设2),进而导出分布函数的表达式:
上面谈到了,即便是孤立系统也会有一定的能量弥散,而且为了方便求解,一般把分布函数扩展为能量在$E$和$E+\Delta E$之间的球壳:
这就是等概率原理。$C$是一个归一化常数,对于量子情况,有
对于经典情况,如果理解为量子统计的经典极限:
对于假设2,必须指出的是,等概率原理仅限于等能量面(球壳)上,对于不同能量的等能面,其有额外的权重$e^{-\beta E}$。
平衡态要求分布函数不随时间变化,但没有指定等能面上分布函数的形状,所以并不冲突。可以从细致平衡原理导出等概率原理:平衡态下从$i$态向$j$态的跃迁概率与从$j$态向$i$态的跃迁概率相等,即:
同时,平衡态要求$i$态的概率不随时间变化,即:
这说明在平衡态下,所有的等能面上的态的概率是相同的。
微正则系综的热力学公式
现在推导热力学公式,此前我们通过让两个热源接触来导出温度的定义,现在我们做同样的事情:假设两个孤立系统$A_1$和$A_2$组成复合系统$A$,复合系统的能量为$E=E_1+E_2$,微观状态数为$\Omega=\Omega_1\Omega_2$,则
由等概率原理,可知当粒子数极大的时候,系统趋向于最概然情况,即$\Omega$取极大值。
从这一步就可略知一二了:微观状态数和熵都有对应的极值原理,不过前者是乘算,后者是加算:
这很自然启发我们对微观状态数取对数得到熵。
对其求导数:
注意到$\frac{\partial E_2}{\partial E_1}=-1$,得到
也就是说热平衡的时候有一个量是守恒的(这不就是热零嘛)。考虑热力学结果$\left(\dfrac{\partial S}{\partial U}\right)_{N,V}=\dfrac1T$,定义该守恒量为:
比较后得到:
同样的道理,如果可以交换粒子和体积,可以分别对$N,V$求导得到
这和之前的热动平衡条件相当。
解题指南:微正则系综的特性函数为$S=k\ln \Omega(N,U,V)$,通过特性函数求出热力学量
所以:
- $\dfrac1T=\dfrac{\partial S}{\partial E}$
- $\dfrac pT=\dfrac{\partial S}{\partial V}$
- $\dfrac {-\mu}T=\dfrac{\partial S}{\partial N}$
所有能量守恒的孤立系统都可以用微正则系综来描述,典型的问题有单原子理想气体、爱因斯坦的固体热容理论、橡胶带模型。由于需要计算微观状态数,这一类问题往往希望条件比较简单,或是对象较少。
单原子经典理想气体
微正则系综,根据等概率原理,考虑粒子的全同性后,微观状态数为:
我们利用 近独立粒子的最概然分布 中的结论:
代入$n=3N$,得到总的微观状态数:
理想气体的熵为:
斯特林近似后:
后两项在热力学极限下可以忽略:
这说明熵是一个广延量,且壳层的宽度不影响熵的数值。
通过上式反解得到能量:
利用特性函数,得到基本热力学函数:
即
代入熵的方程:
这和热力学的结果 热力学基本方程 是定性一致的:
爱因斯坦的固体热容理论
也可以通过分解每个简正模为一个正则系综来求解,思路见 玻尔兹曼统计 。
爱因斯坦的固体热容理论认为所有简正模的振动频率一样,为$\omega_0$,区别在于不同的简正模的量子数不同。平移能量让基态能量为0,该问题等价于让$U/\hbar\omega_0$个总量子数分配在$3N$个简正模上。这样,所有可能的微观状态数为:
通过斯特林近似得到:
这和用正则系综方法算出的表达式是完全相同的(正则系综没有平移零点能,用$U’$表示):
正则系综
正则系综的热力学公式
在微正则系综的计算中,我们已经明显感受到给定能量下计算微观状态数的困难。甚至对于某些不理想的系统,仅仅是计算微观状态数就是不可能的。为了解决这个问题,可以去除能量约束条件,考虑一个具有确定的粒子数$N$、体积$V$和温度$T$的系统。这样的系统称为正则系综。
由于温度确定,可以把正则系综想象成一个微正则系综劈成两半,一个小的是我们要研究的正则系综,另一个虽然也是正则系综,但是由于它的体积和粒子数都很大,所以可以看成是一个热源,他的能量很大,几乎不会因为和小的正则系综交换能量而改变:
依据等概率原理,我们认为正则系综上处于某一分布的概率正比于其上微观状态的数目:
由于热源的能量很大,可以取对数泰勒展开:
所以
这里的$Z$称为配分函数:
经典极限:
解题指南:写出各量子态的能量$E_s$,求出量子或经典情况的表达式$E(q,p)$,进而求得配分函数的表达式。正则系综的特性函数为$F=-k\ln Z(N,V,T)$,利用自由能这一特性函数求出其他热力学量:
所以:
- $S=-\frac{\partial F}{\partial T}$
- $p=-\frac{\partial F}{\partial V}$
- $\mu=\frac{\partial F}{\partial N}$
相比于微正则系综以熵为最大化函数,正则系综以自由能$F=U-TS$为最大化函数。
对于正则系综,能量有涨落,我们可以简单计算出能量分布的方差:
所以相对涨落为:
正则系综的便利性
在利用微正则系综讨论理想气体的时候,我们就注意到了这种方法相比于玻尔兹曼统计方法的繁琐。然而,如果你直接使用正则系综,那么也不可避免产生同样的情况:
但是经过一番计算,你发现了他其实和玻尔兹曼统计中单原子配分函数有着密切的联系:
必须要强调两点:
- 当我们不适用经典表达式(积分)进行研究,而是使用量子表达式(求和)的时候,由于已经考虑量子的全同性原理,则不需要再除以$N!$:
- 在经典极限下,依然需要考虑粒子的全同性,因此在求解时仍然需要除以$N!$。此前的积分表达式$Z=\int_0^\infty \frac{N(E)}{N!} e^{-\beta E}dE$其实是非简并条件近似后的表达式,如果不使用非简并条件近似,则需要考虑能级内的交换数:只是因为非简并条件下,大部分粒子$n_i=0,1\Rightarrow n_i!= 1$,所以可以忽略。
正则系综和微正则系综的联系
求出理想气体的配分函数后:
可以求出自由能:
而根据微正则系综的配分函数:
算出的熵为:
对他们分别求导,得到:
可以看到,这两个方程并不等价,说明正则系综和微正则系综的联系并不是简单的等价关系。实际上,只有满足热力学极限$N\to\infty$时,正则系综和微正则系综才是等价的(这一点在用微正则系综讨论理想气体的时候就提到了)。
对这个关系有一个形式上的证明,参见有丘直方的知乎回答。对其整理如下:
记$I=T^{-1}$。如果正则系综和微正则系综是等价的,那么$E=F’(I)$和$I=S’(E)$应该是同一个方程。然而,由于正则系综的配分函数是微正则系综的配分函数的Laplace变换:
这通常会带来阶的不同,进而导致取对数求导后的系数不同。表述热力学极限可以表述微,将广延量乘以$\lambda$,再令$\lambda\to\infty$。此时根据Laplace方法(见 基础的数理化知识 )可以算出:
进而求得自由能的导数:
其中:
所以:
这说明正则系综和微正则系综在热力学极限下是等价的。
实际气体
实际气体的能量表达式为:
后项是粒子间的相互作用势能,共有$N(N-1)/2\approx \dfrac {N^2}2$项。这时候,如果还坚持用微正则系综求解就得不偿失了,那是我们还需要引入$3N$个位置坐标,且位置空间的分布不像动量空间均匀。这时候用正则系综更合适。
先求出配分函数:
能量可以分离为坐标和动量项,其中动量项处理为:
坐标项保留成积分形式,称为位形积分:
配分函数写为:
定义函数$f_{ij}=e^{-\beta\phi(r_{ij})}-1$,则位形积分可以写为:
保留前两项:
计算得到:
气体的压强为:
即:
考虑Lennard-Jones势:
得到:
或者范德瓦尔斯方程:
德拜的固体热容理论
固体中原子间存在很强的相互作用,将原子间相互作用展开到二级:
当原子位于平衡位置的时候,$\frac{\partial \phi}{\partial \xi_i}=0$,所以能量表达式为:
可以转化为二次型的形式:
量子力学理论指出:
配分函数为:
固体中存在横波和纵波,其中横波有两种形式:
态密度为:
记:
存在积分极限$\omega_D$使得:
解得:
这就是德拜频谱。
计算内能:
代入德拜频谱:
当$T\ll \omega_D$时,可以近似为:
相比于爱因斯坦的固体热容理论,德拜理论考虑了简正模,在极低温的时候所有原子不会被同时冻结,所以收敛较慢,符合实际。
巨正则系综
和大热源和大粒子源接触平衡的系统以化学势,体积和温度为独立变量,叫做巨正则系综。
同样的方法,可以得到
这里先对粒子数求和,对于确定的粒子数,不同的能级分布带来不同的$E_s$,所以还要对能级分布求和。我们可以推导以下等效表达式(对于近独立系统):
这说明巨配分函数可以看成每个能级的“子巨配分函数”之积。
经典极限:
对于玻色统计,我们用上式推导出当时直接给出的式子:
考虑玻色系统任一态的粒子数无限制,其子巨配分函数:
自然得到上式。注意这里区分两个概念:能级的简并数$\omega_l$和一个能级上粒子的数目$n$。简并的两个能级并不需要拥有同样的粒子数,不过上面的证明先计算能级,两个相同的能级自然就可以乘在一起。
对于费米统计:
考虑费米系统的粒子数为0或1,其子巨配分函数:
自然得到上式。
巨配分函数还有第二种表达方式:
对于没有相互作用的粒子,还可以再次简化:
所以
吸附现象
点吸附(0维吸附)
吸附表面有$N_0$个吸附中心,被吸附的气体能量为$-\epsilon_0$,求吸附率。
巨正则系综的配分函数为:
被吸附粒子的平均数为:
所以吸附率为:
再考虑热动平衡和化学势平衡:
代入即可。
面吸附(2维吸附)
被吸附的粒子可以在二维表面上做二维运动,能量为$\frac{p^2}{2m}-\epsilon_0$,求吸附粒子的面密度。
巨正则系综的配分函数为:
所以面密度为:
代入化学势平衡:
表格
| 微正则系综 | 正则系综 | 巨正则系综 | |
|---|---|---|---|
| 系统 | 孤立系统(无粒子能量交换) | 闭合系统(有能量交换) | 开放系统(无能量交换) |
| 独立状态变量 | $N,E,V$ | $N,V,T$ | $\mu,V,T$ |
| 分布概率 | $\rho=\frac{1}{\Omega}$ | $\rho=\frac{\omega_l}{Z}e^{-\beta E}$ | $\rho=\frac{1}{\Xi}e^{-\alpha N-\beta E}$ |
| 配分函数 | $\Omega$ | $Z=\sum \omega_l e^{-\beta E}$ | $\Xi=\sum_{N=0}^\infty\sum_s e^{-\alpha N-\beta E_s}$ |
| 配分函数的经典极限 | $\Omega=\frac{1}{h^{Nr}N!}\int_{E\leq H(q,p)\leq E+\Delta E}d\Omega$ | $Z=\frac{1}{N!h^{Nr}}\int e^{-\beta E(q,p)}d\Omega$ | $\Xi=\sum_{N=0}^\infty\frac{e^{-\alpha N}}{N!h^{Nr}}\int e^{-\beta E(q,p)}d\Omega$ |
| 粒子数 | $N$ | $N$ | $N=-\frac{\partial \ln \Xi}{\partial \alpha}$ |
| 内能 | $U$ | $U=-\frac{\partial \ln Z}{\partial \beta}$ | $U=-\frac{\partial \ln \Xi}{\partial \beta}$ |
| 外力 | ${Y}=-\frac1\beta\frac{\partial \ln \Omega}{\partial y}$ | $Y=-\frac1\beta\frac{\partial \ln Z}{\partial y}$ | $Y=-\frac1\beta\frac{\partial \ln \Xi}{\partial y}$ |
| 化学势 | $\mu=-\frac1\beta\frac{\partial \ln \Omega}{\partial N}$ | $\mu=-\frac1\beta\frac{\partial \ln Z}{\partial N}$ | $\mu$ |
| 熵 | $S=k\ln \Omega$ | $S=k(\ln Z+\beta U)$ | $S=k(\ln \Xi+\alpha N+\beta U)$ |
| 基本热力学关系 | $S=k\ln\Omega$ | $F=-kT\ln Z$ | $J=-kT\ln\Xi$ |
| 特性函数法 | None | $S=-(\frac{\partial F}{\partial T})_{V,N}$ $p=-(\frac{\partial F}{\partial V})_{T,N}$ $\mu=(\frac{\partial F}{\partial N})_{T,V}$ $U=F+TS$ |
$S=-(\frac{\partial J}{\partial T})_{V,\mu}$ $p=-(\frac{\partial J}{\partial V})_{T,\mu}$ $N=-(\frac{\partial J}{\partial \mu})_{T,V}$ |
正则系综和巨正则系综给出一样的物理信息,不过是从不同的自变量,即特性函数出发。处理问题前可以根据给出的条件进行选择。
注意,如果是满足经典极限条件的玻色或费米系统,需要先修正熵的表达式,再求出化学势。